Abstract: Amyotrophic lateral sclerosis(ALS) is the destructive neurodegenerative disease, resulting in the gradual decrease of motoneurons controlling muscle movement in the brain and spinal cord. With the loss of neurons, the muscle becomes weak and atrophic. Movement ability, language expression, eating and breathing of individuals will be gradually damaged, finally resulting in the death. Pathological features of ALS are associated injuries of upper and lower motoneurons. Researches on pathophysiological mechanisms are important for developing new drugs of ALS treatment.
Keywords: Amyotrophic Lateral Sclerosis, Neurodegenerative Disorder, Motor Neurone Disease, Neuromuscular Disorders
1. Causes of ALS
Exact causes of amyotrophic lateral sclerosis(ALS) still remain unclear. But researches show various factors may be involved, including genetic factors, environmental factors and life style etc. About 10% ALS patients have family history. Some known associated genes include SOD1, C9orf72, TARDBP and FUS. The mutation of these genes may cause the incorrect protein folding and aggregation, and affects cell function by resulting in damage and death of motoneurons.
2. Clinical Symptoms
There are various ALS clinical symptoms. This disease affects motoneurons controlling muscle. The development of ALS is usually rapid and irreversible. Symptoms will gradually get worse over time. Typical symptoms include muscle weakness and myatrophy, spasm and convulsion, language and deglutition disorders, respiratory problems and altered reflexes. Patients may suffer from gait disturbances, cognitive and behavioral changes(e.g. frontotemporal lobar dementia), occasional paresthesia, emotional lability and pain.
3. Pathogenesis of ALS
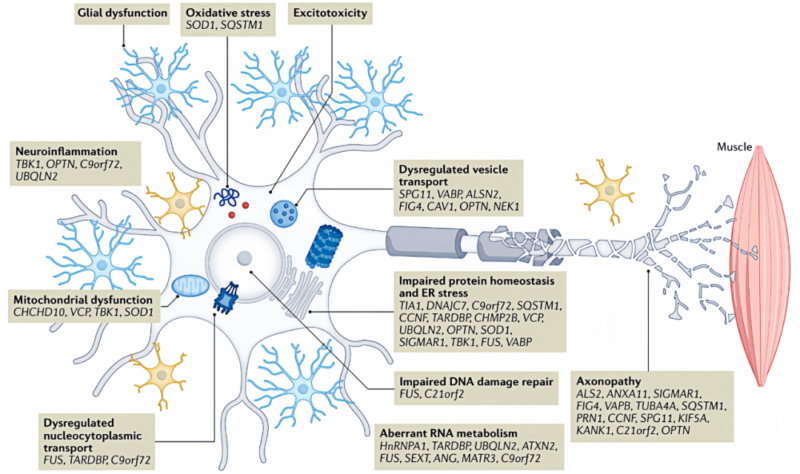
Pathogenesis of ALS is involved in inheritance, oxidative stress, excitotoxicity, neuroinflammation, mitochondrial dysfunction, DNA and RNA injury, damaged protein homeostasis etc.
3.1. Genetic Structure
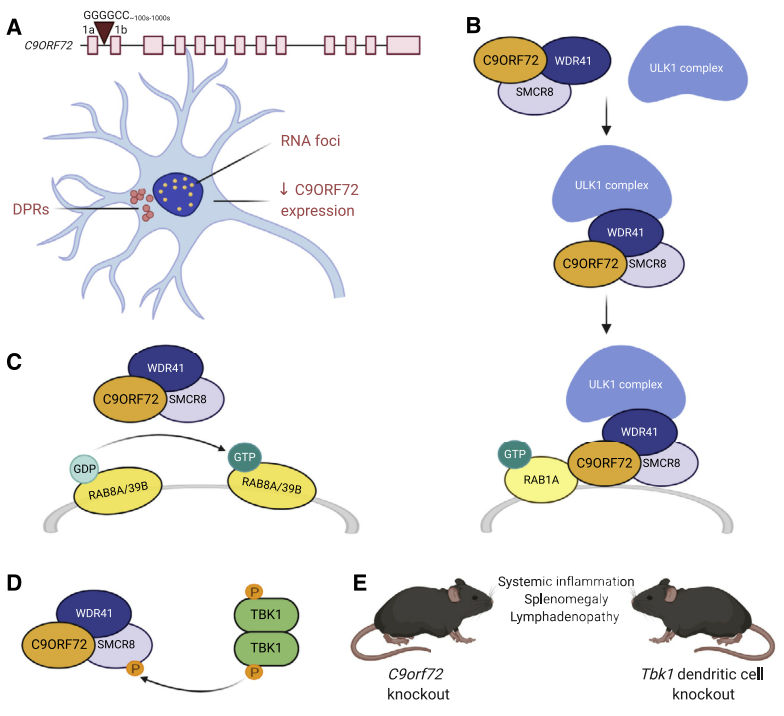
About 5-10% ALS patients have familial predisposition, especially for autosomal dominant inheritance. Genetic factors are key causes of ALS, but only 60-70% ALS familial etiologies are clear. Over 30 genes are related to ALS. Familial ALS cases caused by C9ORF72, SOD1, TARDBP and FUS are 70%. The repeat amplification GGGGCC of C9ORF72 gene is the most common genetic reason for ALS. About 34% such ALS cases are found in European population. The mutation causes the disease by RNA toxicity, DPR toxicity(dipeptide repeat proteins) or functional deficiency of C9ORF72. C9ORF72 is involved in the autophagy process, forms the complex together with SMCR8 and WDR41, and regulates ULK1 complex as well as Rab GTP enzymatic activity. Knockout can cause immune system disorders. Inhibition of STING signaling pathway can relief the symptoms. Besides, C9ORF72 also affects TFEB, and also regulates autophagy and lysosomal function. It's important to remove protein aggregates in the cell.
3.2. Oxidative Stress
Oxidative stress plays an important role in ALS incidence, and is featured with imbalance of glutathione, Nrf2 and antioxidant response. Abnormal aggregation of TDP-43 is closely related to ALS development. Oxidative stress promotes acetylation and phosphorylation, affects RNA binding and enhances nerve injury. Besides, oxidative stress changes the distribution of TDP-43 and FUS and interferes with RNA processing. Up-regulation of cystine/glutamate antiporter increases the release of glutamate by the astrocyte, and also improves the excitotoxicity for motoneurons. These processes result in mitochondrial dysfunction and further oxidative stress.
3.3. Excitotoxicity
In ALS, over activation of postsynaptic glutamate receptor results in excitotoxicity and neuronal damage by increasing intracellular calcium level. In the ALS model, the specificity of motoneurons shows calcium permeability, increase of AMPA receptor and decrease of calcium buffering proteins. The sensitivity of excitotoxicity is enhanced. The mutation of C9orf72 also increases the sensitivity in human induced pluripotent stem cells. Regulation of metabotropic glutamate receptors decreases the release of glutamate and promotes the generation of neurotrophic factors. This may be the new treatment strategy for ALS.
3.4. Neuroinflammation
Neuroinflammation is a typical pathological feature of ALS. The research shows co-culture for astrocyte induced from ALS patients' fibroblast and motoneuron will reveal the toxicity for neurons. Microglia has double roles in nervous system: M1 microglia promotes inflammatory response(neurotoxicity). M2 microglia tends to be anti-inflammation and protecting nerves(neuroprotective effect). The ALS model for SOD1 transgenic mouse observes the protective M2 microglia gradually changes into pro-inflammatory M1 microglia. Besides, ALS patients' peripheral blood monocyte is more easily activated, and tends to differentiate into M1 microglia. Activation of NLRP3 inflammasomes is thought to be one of the key factors of neuroinflammation in ALS. Inhibiting activation of NLRP3 inflammasome may improve ALS symptoms by reducing activated microglia induced-neuroinflammation. This is a new potential treatment strategy.
3.5. Mitochondrial Dysfunction
Mitochondrial dysfunction is the key ALS pathogenesis, featured by energy dysmetabolism, increased active oxygen, axonal transport interruption and structural dynamics changes. The final result is apoptosis. The mutation of SOD1 aggregates in mitochondrial intermembrane space to decrease activity of electron transfer chain. poly-GR produced by the mutation of C9orf72 binds to ATP5A1 and accelerates ubiquitination and degradation. TDP-43 maintains mitochondrial homeostasis by regulating mitochondrial transcription. These changes accelerate mitochondrial dysfunction in ALS.
3.6. DNA and RNA Damage
Level of oxidized deoxyguanosine in ALS patients' central nervous system increases and DNA is damaged. Apurinic or apyrimidinic sites increase. DNA damage response(DDR) is activated. TDP-43 regulates the signal of DDR. The deficiency is related to DDR. TDP-43 is involved in repair of non-homologous end joining. The mutation of TARDBP inhibits the process and results in DNA damage. The deficiency of TDP-43 also increases R-loop formation and genomic instability. The mutation of FUS and NEK1 also affects DNA repair. Besides, the mutation of C9orf72 causes RNA accumulation, interfere with metabolic processes like RNA splicing and results in damaged nucleocytoplasmic transport. Inhibiting nuclear export of TDP-43 can reduce neuron death.
4. Detection Methods
Clinical manifestations of ALS individuals are greatly different. There are various early symptoms. It's difficult to diagnose due to lack of specific biomarkers. Various detection methods are required, including nerve electrophysiological examination(e.g. nerve conduction testing and needle electromyography) for evaluating neural and neuromuscular electrical activities; magnetic resonance imaging(MRI) for eliminating other possible diseases; blood and urine testing for eliminating heavy metal intoxication, autoimmune diseases or vitamin deficiency; CSF analysis for eliminating infection or other nervous system diseases; gene detection of patients with familial history to confirm genetic type. In some cases, comprehensive evaluations for eliminating other possibilities should be conducted for accurate diagnosis, like biopsy for muscles and nerves, pulmonary function test, ultrasound examination and determination of neurofilament light chain level in cerebrospinal fluid and serum etc.
5. Recommended Products
|
Proteins |
|||
|
Cat.No |
Product Name |
Mol. Weight |
Host |
|
Recombinant Human SOD1 |
16.7 kDa |
E.Coli |
|
|
Recombinant Human C9orf72 |
42.8 kDa |
E.Coli |
|
|
Recombinant Human TARDBP |
28.5 kDa |
E.Coli |
|
|
Antibodies |
|||
|
Cat.No |
Product Name |
Tested Application |
Antibody Type |
|
SOD1 antibody |
ELISA, WB, IHC, IP, IF |
Rabbit pAb |
|
|
C9orf72 antibody |
ELISA, WB, IHC, IF, IP |
Rabbit pAb |
|
|
TARDBP antibody |
ELISA, IP, FC, WB, IHC |
Rabbit mAb |
|
|
ELISA Kits |
|||
|
Cat.No |
Product Name |
Range |
Sensitivity |
|
Human SOD1 ELISA Kit |
0.781-50ng/ml (10000IU/mg) |
0.469ng/ml |
|
|
Mouse SOD1 ELISA Kit |
0.781-50ng/ml |
0.469ng/ml |
|
|
Rat SOD1 ELISA Kit |
0.781-50ng/ml |
0.469ng/ml |
|
|
Human FUS ELISA Kit |
78.125-5000pg/ml |
46.875pg/ml |
|
|
Mouse TDP43 ELISA Kit |
0.313-20ng/ml |
0.188ng/ml |
|
|
Human TDP43 QuickTest ELISA Kit |
0.313-20ng/ml |
0.188ng/ml |
|
|
Human NGFR QuickTest ELISA Kit |
1.563-100ng/ml |
0.938ng/ml |
|
REFERENCES
[1]Advances in molecular pathology, diagnosis, and treatment of amyotrophic lateral sclerosis, PMID: 37890889.
[2]Drug discovery and amyotrophic lateral sclerosis: Emerging challenges and therapeutic opportunities, PMID: 36402404.